lecture_notes:04-08-2015
Table of Contents
De novo assembly II
Guest lecturer: Stefan Prost, stefan.prost@berkley.edu
Illumina paired-end sequencing libraries
- MiSeq has 300 bp reads
- Paired ends read from both directions, so you get one read for each end (may or may not overlap depending on molecule and read size)
=====> end_1 ________________________ end_2 <=====
- Problem: not really sufficient for repetitive regions
- ends can't be very far apart because Illumina can't handle big molecules
- not enough info for scaffolding
Illumina mate-pair sequencing libraries
- Idea: get paired reads that are much farther away (for more scaffolding info)
- Basically, the same idea as paired-ends, except the middle section is missing and the ends are oriented the opposite way.
<===== end_1 ________________________ end_2 =====>
- Genomic DNA > Fragment (2-5 kb) > biotinylate ends > Circularize > Fragment (400-600 bp) > Enrich biotinylated fragments > Ligate adaptors > …
- Cut DNA, attach a biotin tag to both ends of the target molecule
- Circularize target molecule
- This step is hard
- Circularized molecule is fragmented, fragment with the biotin tag (a.k.a the ends of the original target stuck to each other backwards, with the tags in between) is enriched
- Then end repair, A-tailing, adapters added, amplification, sequencing
- Dependent on inferring insert size (can be tricky)
- Most companies can get you 8 kb inserts, with skill you can get up to 20 kb
- Complicated process, weird stuff can happen in between
- Important difference between paired ends and mate-pairs: ends are oriented the opposite way.
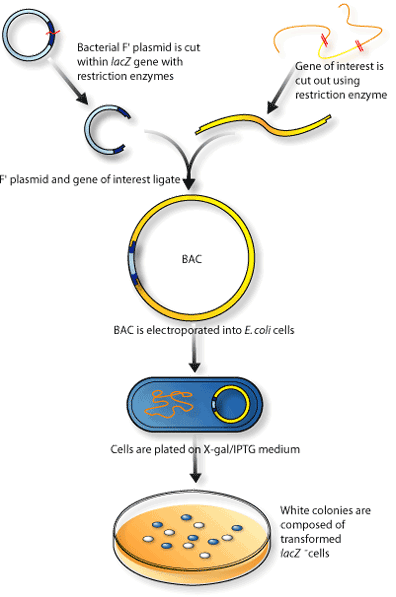
BAC (Bacterial Artificial Chromosome) and fosmid libraries
- Uncommon and expensive, but the gold standard
- Bacterial F-plasmid takes< 40 kb insert size
- Fragment query DNA > DNA into BAC > Transform E. coli with BAC > ~300 kb long reads
{kind=link}
Read quality assessment
- Base quality: Phred scores reported by sequencer.
- Fastq files: fasta files, plus encoded phred scores
- Need to know if your file has phred33 or phred64 encoding
- Quality for each individual base is not the whole story, the context matters to the signal processing too
- Reads decrease in quality further down the read
- Depending on the assembler, you might need to trim lower quality reads off the end. Others want untrimmed data
- Pacific Bio doesn’t have GC|AT bias
Tools
- FastQC (Most popular tool to tell you about the read library)
- FastQC reported an issue with our data with kmer count (related to adapter content)
- This needs to be checked out and diagnosed!
- Preqc
- Estimates how difficult the assembly will be
Estimating genome size from read data
G = (pn(1-k+1))/(λ_k) G = Genome size pn = proportion of correct reads k = kmer length λ_k= mode of the k-kmer count histogram Simpson 2013, arXiv
- To estimate genome size need to know:i) total number of reads; ii) length of reads; and iii) kmer distribution
Error correction
- High amount of small kmers are usually errors
Simulated contig length in the k-de Brujin graph can estimate the best kmer to use for assembly. Based on contig length N50
You could leave a comment if you were logged in.
lecture_notes/04-08-2015.txt · Last modified: 2015/04/17 22:34 by sihussai
Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Noncommercial-Share Alike 3.0 Unported
