De novo assembly II
Guest lecturer: Stefan Prost, stefan.prost@berkley.edu
Illumina paired-end sequencing libraries
=====> end_1
________________________
end_2 <=====
Illumina mate-pair sequencing libraries
Idea: get paired reads that are much farther away (for more scaffolding info)
Basically, the same idea as paired-ends, except the middle section is missing and the ends are oriented the opposite way.
<===== end_1
________________________
end_2 =====>
Genomic DNA > Fragment (2-5 kb) > biotinylate ends > Circularize > Fragment (400-600 bp) > Enrich biotinylated fragments > Ligate adaptors > …
Cut DNA, attach a biotin tag to both ends of the target molecule
Circularize target molecule
Circularized molecule is fragmented, fragment with the biotin tag (a.k.a the ends of the original target stuck to each other backwards, with the tags in between) is enriched
Then end repair, A-tailing, adapters added, amplification, sequencing
Dependent on inferring insert size (can be tricky)
Most companies can get you 8 kb inserts, with skill you can get up to 20 kb
Complicated process, weird stuff can happen in between
Important difference between paired ends and mate-pairs: ends are oriented the opposite way.
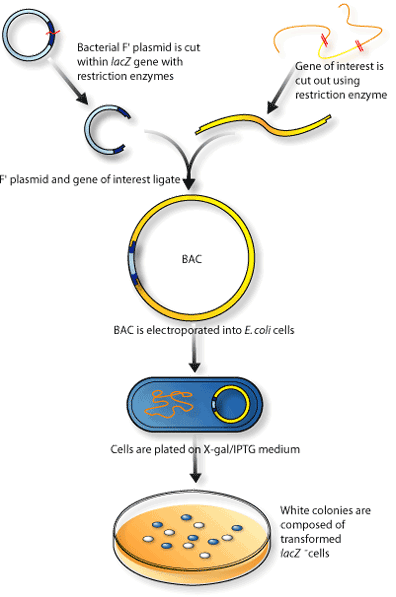
BAC (Bacterial Artificial Chromosome) and fosmid libraries
Uncommon and expensive, but the gold standard
Bacterial F-plasmid takes< 40 kb insert size
Fragment query DNA > DNA into BAC > Transform E. coli with BAC > ~300 kb long reads
-
Read quality assessment
Base quality: Phred scores reported by sequencer.
Fastq files: fasta files, plus encoded phred scores
Quality for each individual base is not the whole story, the context matters to the signal processing too
Reads decrease in quality further down the read
Pacific Bio doesn’t have GC|AT bias
Estimating genome size from read data
G = (pn(1-k+1))/(λ_k)
G = Genome size
pn = proportion of correct reads
k = kmer length
λ_k= mode of the k-kmer count histogram
Simpson 2013, arXiv
Error correction
Simulated contig length in the k-de Brujin graph can estimate the best kmer to use for assembly. Based on contig length N50
{kind=link}